Study design
Design
The Dutch ICH Surgery Trial (NCT05460793) is a multicenter phase III, prospective, randomized, open, blinded endpoint (PROBE) clinical trial. Patients will be recruited in 11 neurosurgical centers in the Netherlands. In addition, local investigators in ~33 general hospitals without facilities for intracranial neurosurgery but with experience in clinical trials in stroke, will be part of the study group and refer patients for inclusion. The study will run for 4 years, which includes a 3-year inclusion period with a follow-up period of 12 months.Population
We will include 600 patients with a spontaneous, supratentorial intracerebral hemorrhage of 18 years and older with a minimal hematoma volume of 10 mL and a NIHSS of 2 or higher.
Patients with an aneurysm, arteriovenous malformation, dural arteriovenous fistula, or cerebral
venous sinus thrombosis as cause of their ICH will be excluded based on the admission CT
angiography. Patients with a known tumor or cavernoma will also be excluded.
For the DIST-INFLAME substudy, we will include 200 patients; 100 randomized to intervention and 100 randomized to standard medical management.
In- and exclusion criteria
| Inclusion checklist |
| Age ≥18 years |
| NIH Stroke Scale score ≥ 2 |
| Supratentorial ICH confirmed by non-contrast CT, without a CT-angiography confirmed causative vascular lesion (e.g., aneurysm, arteriovenous malformation, dural arteriovenous fistula, cerebral venous sinus thrombosis) or other known underlying lesion (e.g., tumor, cavernoma) |
| Minimal ICH volume of 10 mL |
| Intervention can be started within 8 hours of symptom onset |
| Written informed consent (deferred) |
| Exclusion checklist |
| Pre-stroke mRS ≥3 |
| ICH-GS score ≥11 |
| Hemorrhage due to hemorrhagic transformation of an infarct |
| Untreated coagulation abnormalities, including INR > 1.3 (point of care measurement allowed) and treatment with thrombin or oral factor Xa antagonists. |
| Moribund (e.g., coning, bilateral dilated unresponsive pupils), or progressively deteriorating clinical course with imminent death |
| Pregnancy |
| DIST-INFLAME sub-study: patients that use immunosuppressive or immune-modulating medication |
Intervention
Minimally invasive endoscopy-guided surgery within 8 hours of symptom onset, in addition to standard medical management. The investigational product is: a device for minimally invasive, endoscopy-guided hematoma removal. Currently, the second generation Penumbra device, the Artemis system, is available and CE approved. When other devices will become available, they may be used when they are CE approved and deemed admissible by the steering committee.
Main study parameters/endpoints
The primary outcome parameter will be the modified Rankin scale (mRS) score at 180 days. This categorical scale measures functional outcome with scores ranging from 0 (no symptoms) to 6 (death). The treatment effect will be estimated with ordinal logistic regression analysis as common odds ratio, adjusted for prespecified prognostic factors. The adjusted common odds ratio will measure the likelihood that minimally invasive endoscopy-guided surgery will lead to lower mRS scores as compared to standard medical management alone.Secondary outcomes will include: the score on the mRS at 90 and 365 days; favorable outcome (defined as a mRS 0-2 and 0-3) and all other possible dichotomizations of the mRS at 90, 180 and 365 days; NIHSS at day 6 (±1 day); death, Barthel Index, EuroQol-5D-5L, SS-QOL, iMCQ, iPCQ and iVICQ at 90, 180 and 365 days.
Safety outcomes will be death within 24 hours, at 7 and at 30 days and procedure-related complications within 7 days.
Technical effectiveness outcomes will be percentage volume reduction based on the baseline CT and CT at 24 hours (± 6 hours), percentage of participants with clot volume reduction ≥70%, and ≥80%, and with remaining clot volume ≤10mL, and ≤15mL, and conversion to craniotomy.
In DIST-INFLAME, outcomes will include perihematomal edema at 6 days (±1 day), functional outcome at 180 days and immune and metabolomic profiles at 3 (± 12 hours) and 6 days (±1 day).
Informed consent
In this study, we will defer written consent until after the treatment, or after randomization forpatients in the control arm. We will strive to obtain consent as soon as possible but when deemed reasonable and appropriate, preferably within 24 hours. When the patient is not competent, the investigator will search for a legal representative available.
Flowchart of study procedures
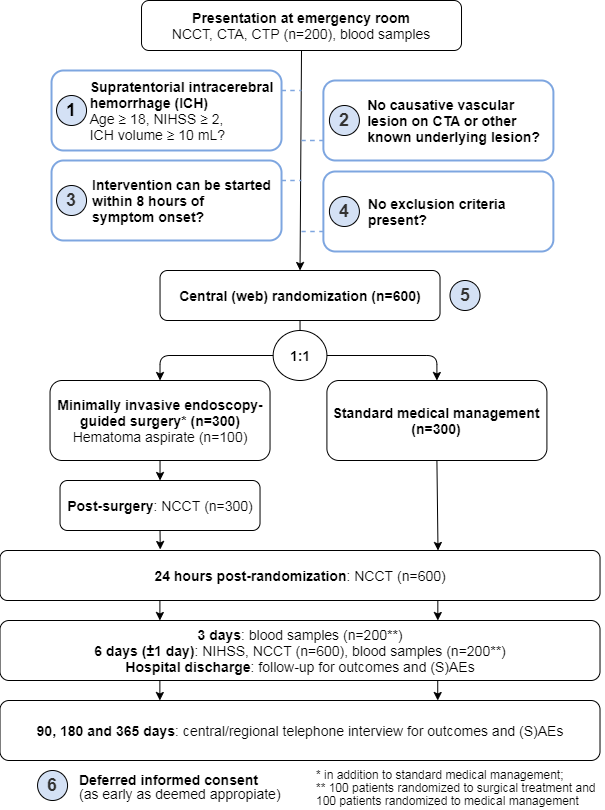
CTA: Computed tomography angiogram; CTP: CT Perfusion; ICH: intracerebral hemorrhage; NCCT: Non-contrast computed tomography; NIHSS: National Institutes of Health Stroke Scale; (S)AEs: (Serious) Adverse Events.